

ورود به عرصه تولید تجهیزات پزشکی در ایران، سفری پیچیده و چندوجهی است که در قلب آن، یک سند حیاتی به نام “پروانه ساخت” قرار دارد. این پروانه، صرفاً یک مجوز اداری نیست؛ بلکه تأییدیه رسمی دولت بر توانایی یک تولیدکننده برای تولید مستمر و قابل اتکای تجهیزات پزشکی ایمن و مؤثر است. دریافت این سند، نقطه عطفی است که یک ایده نوآورانه را به یک محصول قانونی و قابل عرضه در بازار سلامت کشور تبدیل میکند و دروازهای برای ورود به اکوسیستم درمانی ایران محسوب میشود.
اهمیت استراتژیک این پروانه فراتر از الزامات قانونی صرف است. این مجوز، پیشنیاز اصلی برای فروش و توزیع قانونی محصولات، شرکت در مناقصات دولتی که بخش بزرگی از بازار را تشکیل میدهند، و ایجاد اعتماد در میان جامعه پزشکی و بیماران است. بدون پروانه ساخت، هرگونه فعالیت تجاری در این حوزه غیرقانونی تلقی شده و محصول تولیدی به عنوان کالای قاچاق شناخته میشود که میتواند منجر به پیگردهای قانونی جدی شود.
در بطن این فرآیند نظارتی دقیق، اصلی بنیادین نهفته است: حفاظت از سلامت عمومی و ایمنی بیمار. هر الزام، هر مدرک و هر بازرسی در مسیر اخذ پروانه ساخت، با هدف به حداقل رساندن خطرات ناشی از تجهیزات پزشکی ناکارآمد یا ناایمن طراحی شده است. این فرآیند، یک ارزیابی جامع نه فقط از یک محصول، بلکه از کل یک سازمان است. مرجع نظارتی، علاوه بر دادههای فنی محصول مانند گزارشهای آزمون و پرونده فنی، به الزامات سازمانی مانند ثبت رسمی شرکت، معرفی مسئول فنی واجد شرایط و مهمتر از همه، پیادهسازی یک سیستم مدیریت کیفیت جامع و دارای گواهینامه ایزو $13485$ توجه ویژهای دارد. این رویکرد نشان میدهد که از دیدگاه رگولاتور، توانایی یک سازمان برای تولید مداوم و تکرارپذیر یک محصول باکیفیت، به اندازه کیفیت خود محصول در یک نقطه زمانی خاص، اهمیت دارد. بنابراین، متقاضیان باید فرآیند اخذ پروانه را نه به عنوان یک وظیفه ثبت محصول، بلکه به عنوان یک پروژه توسعه سازمانی در نظر بگیرند که در آن، ایجاد یک فرهنگ کیفیتمحور از روز اول، کلید موفقیت است.
بخش ۱: چشمانداز نظارتی: حاکمیت و مبانی قانونی
مرجع مرکزی: درک نقش اداره کل تجهیزات و ملزومات پزشکی (IMED)
مرکز ثقل نظارت بر حوزه تجهیزات پزشکی در ایران، “اداره کل تجهیزات و ملزومات پزشکی” است که به اختصار IMED نامیده میشود. این اداره، به عنوان زیرمجموعهای از سازمان غذا و دارو، تنها نهاد حاکمیتی مسئول برای تنظیم و نظارت بر کل چرخه عمر تجهیزات پزشکی در کشور است؛ از ارزیابیهای پیش از ورود به بازار گرفته تا نظارتهای پس از عرضه (Post-Market Surveillance). وبسایت رسمی این اداره به نشانی imed.ir، به عنوان قطب اصلی تمام فعالیتهای نظارتی، شامل سامانههای ثبت درخواست، پایگاه داده محصولات مجاز، دستورالعملها و اطلاعیههای رسمی عمل میکند.
بستر حقوقی: آییننامههای کلیدی حاکم بر تولید تجهیزات پزشکی
فعالیت اداره کل تجهیزات پزشکی بر پایه یک چارچوب قانونی مستحکم استوار است. “آییننامه فعالیت در حوزه تجهیزات پزشکی” به عنوان اصلیترین سند قانونی، اختیارات لازم را به وزارت بهداشت، درمان و آموزش پزشکی برای نظارت بر این حوزه اعطا میکند. این آییننامه و سایر قوانین مرتبط، مبنای حقوقی تمام الزامات، ضوابط و فرآیندهای مربوط به صدور پروانه ساخت را تشکیل میدهند و درک مفاد آن برای هر تولیدکنندهای ضروری است.
اکوسیستم استانداردها: مروری بر $ISO 13485$، GMP و الزامات ملی
تولیدکنندگان تجهیزات پزشکی در ایران ملزم به رعایت سلسله مراتبی از استانداردها هستند که ایمنی و کیفیت را در سطوح مختلف تضمین میکنند:
- ایزو $13485$ ($ISO 13485$): این استاندارد بینالمللی، به عنوان “استاندارد طلایی” برای سیستمهای مدیریت کیفیت (QMS) در صنعت تجهیزات پزشکی شناخته میشود. پیادهسازی و اخذ گواهینامه $ISO 13485$ یک الزام اجباری برای تولیدکنندگان در ایران است و نشاندهنده تعهد سازمان به فرآیندهای کنترلشده و کیفیتمحور است.
- اصول تولید خوب (GMP): مجموعهای از اصول و رویهها است که تضمین میکند محصولات به طور مداوم و مطابق با استانداردهای کیفی تولید و کنترل میشوند. این اصول، شرایط محیطی، تجهیزات، پرسنل و مستندسازی را پوشش میدهد.
- کد ثبت تجهیزات پزشکی (IRC): یک کد شناسایی منحصربهفرد ملی است که پس از تأیید نهایی، به هر وسیله پزشکی مجاز اختصاص مییابد. این کد برای ردیابی، نظارت پس از فروش و دسترسی به بازار ضروری است و بر روی برچسب اصالت کالا درج میشود.
سیستم نظارتی ایران به طور فعال در حال هماهنگسازی خود با استانداردهای بینالمللی، به ویژه اصول نشان CE اروپا است، اما در نهایت، اقتدار ملی خود را حفظ میکند. این رویکرد، یک چالش و در عین حال یک فرصت برای تولیدکنندگان ایجاد میکند. الزامات مربوط به پرونده فنی و الزام به ارائه گواهی CE برای تجهیزات با کلاس خطر بالا، نشان میدهد که IMED از چارچوبهای معتبر جهانی برای ارزیابی کیفیت و ایمنی بهره میبرد. این امر به تولیدکنندگان اجازه میدهد تا با طراحی سیستمها و مستندات خود بر اساس استانداردهای جهانی، علاوه بر کسب مجوز داخلی، مسیر خود را برای ورود به بازارهای صادراتی نیز هموار سازند. با این حال، باید توجه داشت که حتی با داشتن گواهی CE، فرآیند ثبت ملی و اخذ پروانه ساخت از IMED همچنان یک الزام قطعی است؛ گواهی CE میتواند این فرآیند را تسریع کند، اما جایگزین آن نخواهد بود.

بخش ۲: پیشنیازهای بنیادین: آمادهسازی برای یک درخواست موفق
پیش از ورود به مراحل اصلی ثبت محصول، متقاضیان باید مجموعهای از پیشنیازهای حقوقی، فنی و اداری را برآورده سازند. این مراحل، شالوده درخواست را تشکیل میدهند و هرگونه نقص در آنها میتواند کل فرآیند را متوقف کند.
ساختار شرکتی: الزامات قانونی ثبت شرکت تجهیزات پزشکی
اولین و اساسیترین گام، تأسیس یک شخصیت حقوقی رسمی است. متقاضیان باید شرکتی (معمولاً با مسئولیت محدود یا سهامی خاص) را در اداره ثبت شرکتها به ثبت برسانند که در موضوع فعالیت آن به صراحت به “تولید تجهیزات و ملزومات پزشکی” اشاره شده باشد. این شخصیت حقوقی، به عنوان متقاضی رسمی پروانه ساخت شناخته میشود و تمام مسئولیتهای قانونی بر عهده آن خواهد بود.
نقش حیاتی “مسئول فنی”
- معرفی و صلاحیتها: هر واحد تولیدی موظف است یک “مسئول فنی” واجد شرایط را به اداره کل تجهیزات پزشکی معرفی کند. این فرد باید دارای مدرک تحصیلی مرتبط (مانند مهندسی پزشکی، پزشکی، داروسازی یا رشتههای پیراپزشکی) باشد و صلاحیت وی باید به تأیید IMED برسد.
- مسئولیتها: مسئول فنی، از نظر قانونی و فنی، مسئول نظارت بر تمامی جنبههای کیفی فرآیند تولید، انطباق با ضوابط و مقررات، و ارتباط فنی با اداره کل تجهیزات پزشکی است. حضور و تأیید او در تمامی مراحل، از تدوین مستندات تا تولید نهایی، الزامی است.
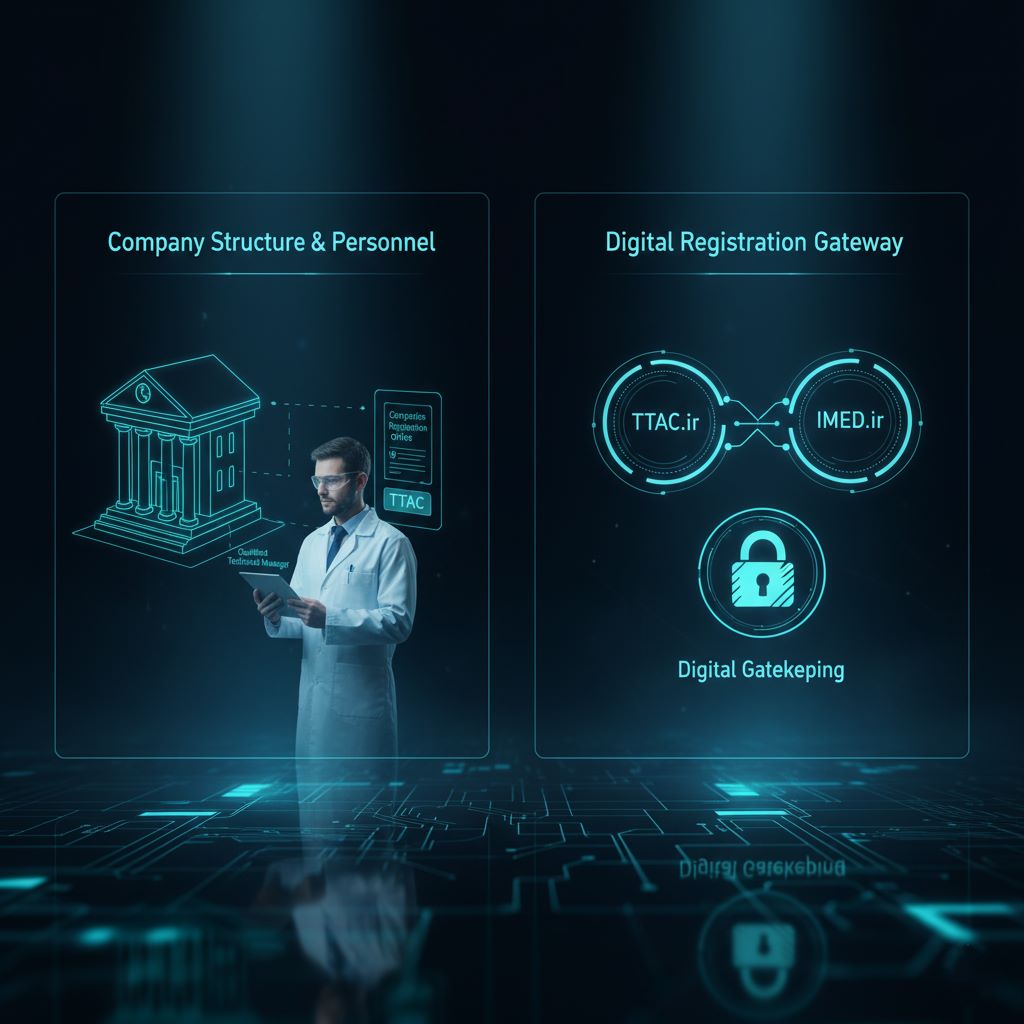
ثبتنام اولیه دیجیتال: پیمایش درگاههای TTAC و IMED
فرآیند درخواست پروانه ساخت کاملاً الکترونیکی است و از طریق دو سامانه مجزا اما مرتبط انجام میشود. این ساختار دو مرحلهای به عنوان یک سیستم “دروازهبانی دیجیتال” عمل میکند تا اطمینان حاصل شود که تنها شرکتهای معتبر و پاسخگو میتوانند فرآیند ثبت محصول را آغاز کنند.
- گام اول: ثبتنام در سامانه TTAC (ttac.ir): شرکت ابتدا باید به عنوان یک شخص حقوقی در سامانه ردیابی، رهگیری و کنترل اصالت فرآوردههای سلامتمحور (TTAC) که متعلق به سازمان غذا و دارو است، ثبتنام کند. در این سامانه، هویت حقوقی شرکت تأیید شده و مسئول فنی معرفیشده نیز ثبت و به پروفایل شرکت متصل میشود. این مرحله، صحتسنجی پاسخگویی قانونی و انسانی شرکت است.
- گام دوم: ثبتنام در سامED (imed.ir): پس از تکمیل موفقیتآمیز ثبتنام در TTAC، شرکت باید پروفایل خود را در سامانه تخصصی اداره کل تجهیزات پزشکی (IMED) ایجاد کند. این سامانه، بستر اصلی برای تمام مراحل بعدی مربوط به ثبت وسیله پزشکی، بارگذاری مستندات فنی و پیگیری پرونده است. این مرحله، به ارزیابی دادههای فنی و محصولمحور اختصاص دارد.
این تفکیک عمدی است و از ارسال درخواستهای ناقص یا غیرجدی به کارشناسان فنی IMED جلوگیری میکند. متقاضیان باید ثبتنام در TTAC را نه یک تشریفات اداری ساده، بلکه ستون اصلی حضور نظارتی خود بدانند، زیرا هرگونه نقص در این مرحله، کل جدول زمانی پروژه را متوقف خواهد کرد.
بخش ۳: ارائه درخواست اصلی: یک رویکرد مرحلهای برای اخذ پروانه
پس از ایجاد زیرساختهای لازم، فرآیند اصلی اخذ پروانه ساخت آغاز میشود که شامل چندین مرحله کلیدی و به هم پیوسته است.
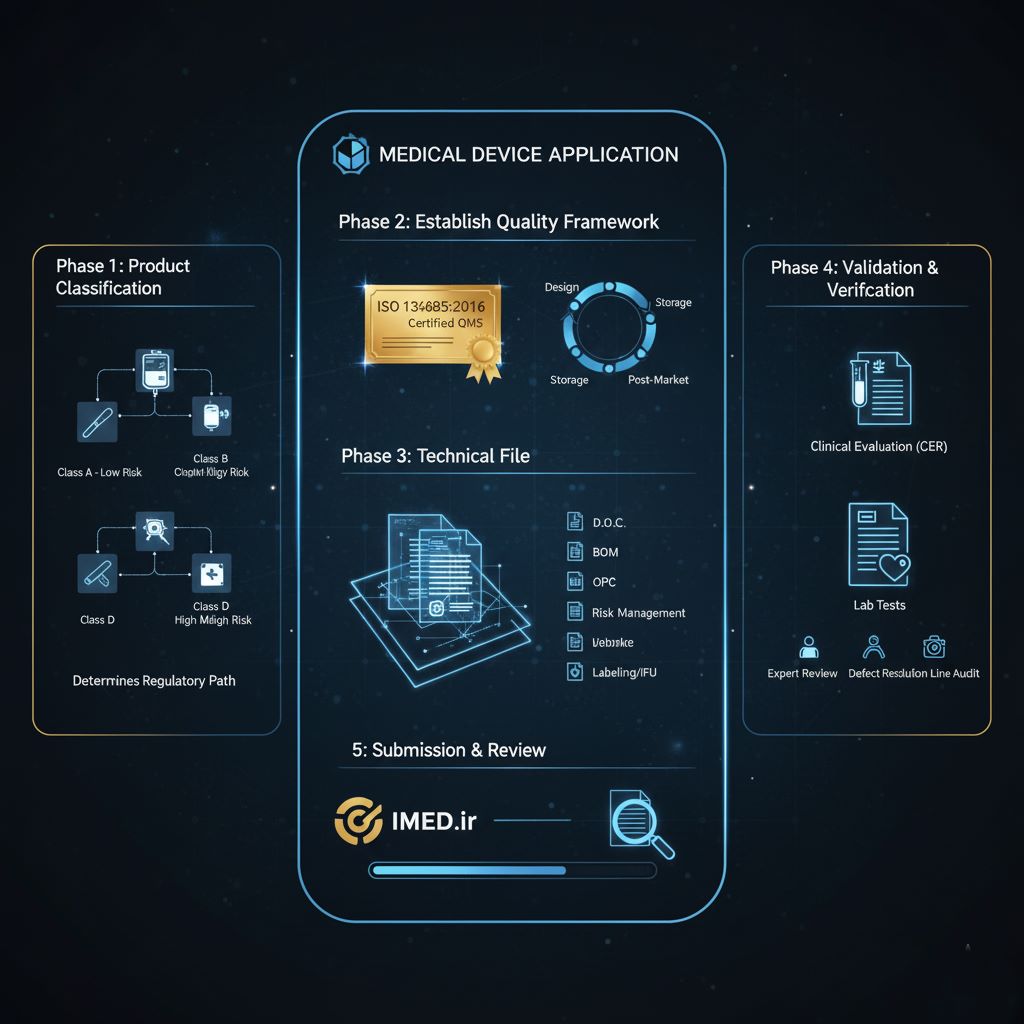
فاز ۱: طبقهبندی محصول – تعیینکننده مسیر شما
- درک کلاسهای خطر: اولین و مهمترین اقدام، تعیین کلاس خطر وسیله پزشکی است. بر اساس پتانسیل خطری که یک وسیله میتواند برای بیمار یا کاربر ایجاد کند، تجهیزات پزشکی به چهار کلاس A (کمترین خطر)، B، C و D (بیشترین خطر) تقسیمبندی میشوند. به عنوان مثال، یک آبسلانگ در کلاس A قرار میگیرد، در حالی که یک ضربانساز قلب (Pacemaker) در کلاس D جای دارد.
- پیامدهای طبقهبندی: کلاس خطر، نقشه راه شما را تعیین میکند. این طبقهبندی، سطح سختگیری نظارتی، عمق مستندات مورد نیاز، و الزامات مربوط به آزمونها و ارزیابیهای بالینی را مشخص مینماید. الزامات برای کلاسهای C و D به مراتب سنگینتر از کلاس A است و اشتباه در تعیین کلاس خطر میتواند منجر به رد کامل درخواست شود.
فاز ۲: ایجاد چارچوب کیفیت
- پیادهسازی یک سیستم مدیریت کیفیت (QMS) قوی: تولیدکننده باید یک QMS جامع را بر اساس الزامات استاندارد $ISO 13485$ ایجاد و پیادهسازی کند. این سیستم باید تمام فرآیندهای مرتبط با چرخه عمر محصول، از طراحی و توسعه تا تولید، انبارش، توزیع و خدمات پس از فروش را پوشش دهد.
- مسیر اخذ گواهینامه $ISO 13485:2016$: این گواهینامه یک نقطه عطف غیرقابل مذاکره است. فرآیند آن شامل انتخاب یک شرکت گواهیدهنده (CB) معتبر، انجام ممیزیهای دقیق و اثبات انطباق کامل با الزامات استاندارد است. برای تجهیزات کلاس A، ممکن است ارائه تعهدنامه یکساله برای اخذ گواهینامه در ابتدای کار پذیرفته شود، اما برای کلاسهای بالاتر، ارائه گواهینامه در زمان ثبت درخواست الزامی است.
فاز ۳: تدوین پرونده فنی – رزومه علمی محصول شما
پرونده فنی (Technical File)، قلب درخواست شما و مجموعهای از مستندات است که ایمنی و عملکرد محصول را به طور علمی اثبات میکند. اجزای اصلی این پرونده عبارتند از:
- اظهارنامه انطباق (D.O.C): یک سند رسمی که توسط مدیرعامل و مسئول فنی امضا میشود و در آن، شرکت تعهد میکند که وسیله پزشکی تولیدی با تمام الزامات اساسی و قوانین مربوطه مطابقت دارد.
- فهرست مواد اولیه (BOM): لیستی جامع از تمام مواد خام، قطعات و تأمینکنندگان آنها که در ساخت وسیله به کار رفتهاند.
- نمودار فرآیند عملیات (OPC): یک فلوچارت دقیق که کل فرآیند تولید را از ورود مواد اولیه تا بستهبندی و عرضه محصول نهایی به تصویر میکشد.
- پرونده مدیریت ریسک (بر اساس $ISO 14971$): مستنداتی که نشان میدهد شرکت یک فرآیند سیستماتیک برای شناسایی، ارزیابی و کنترل خطرات مرتبط با وسیله پزشکی را پیادهسازی کرده است.
- برچسبگذاری و دستورالعمل استفاده (IFU): تمامی برچسبها، بستهبندیها و راهنماهای کاربری باید مطابق با ضوابط ملی طراحی شده و برای بررسی ارائه شوند.
فاز ۴: اعتبارسنجی و صحهگذاری
- انجام آزمونهای آزمایشگاهی: برای اثبات عینی عملکرد و ایمنی محصول، انجام آزمونهای تخصصی در آزمایشگاههای همکار و مورد تأیید اداره کل تجهیزات پزشکی الزامی است. نتایج این آزمونها باید با ادعاهای عملکردی شرکت مطابقت داشته باشد.
- الزامات ارزیابی بالینی: برای تجهیزات با کلاس خطر بالاتر (B, C, D) و فناوریهای نوین، ارائه “گزارش ارزیابی بالینی” (CER) ضروری است. این گزارش شامل بررسی جامع دادههای بالینی برای اثبات ایمنی و کارایی بالینی وسیله در شرایط استفاده واقعی است.
فاز ۵: ارسال نهایی و بررسی در سامانه IMED
- بارگذاری پرونده: پس از تکمیل تمام مستندات، پرونده کامل از طریق سامانه ثبت تجهیزات پزشکی (register.imed.ir) بارگذاری میشود.
- فرآیند بررسی: پس از ارسال، پرونده به یک کارشناس متخصص در اداره کل ارجاع داده میشود. کارشناس مستندات را بررسی کرده و در صورت وجود نقص، موارد را جهت اصلاح (رفع نقص) به شرکت اعلام میکند. در مراحل بعدی، هماهنگیهای لازم برای بازدید و ممیزی از خط تولید نیز توسط کارشناس انجام میشود.
بخش ۴: چکلیست جامع مستندات
یکی از بزرگترین چالشها برای متقاضیان، درک دقیق مستندات مورد نیاز بر اساس کلاس خطر محصول است. ارائه مدارک ناقص یا نادرست، اصلیترین دلیل تأخیر یا رد درخواستها است. جدول زیر به عنوان یک ابزار کاربردی، الزامات مستندسازی را برای کلاسهای خطر مختلف مقایسه میکند و به متقاضیان کمک میکند تا از کامل بودن پرونده خود اطمینان حاصل کنند.
| مدرک/الزام | تجهیزات کلاس A | تجهیزات کلاس B، C و D | نکات و ملاحظات کلیدی |
| مستندات شرکتی و پرسنلی | |||
| مدارک ثبت شرکت | الزامی | الزامی | اثبات هویت حقوقی شرکت. |
| مسئول فنی تأیید شده | الزامی | الزامی | باید صلاحیت وی در سامانه TTAC تأیید شده باشد. |
| سیستم مدیریت کیفیت | |||
| گواهینامه $ISO 13485:2016$ | الزامی (یا تعهدنامه یکساله) | الزامی (در زمان ثبت درخواست) | سنگ بنای QMS. مهلت یکساله برای کلاس A یک تفاوت کلیدی است. |
| پرونده فنی محصول | |||
| اظهارنامه انطباق (D.O.C) | الزامی | الزامی | با امضای مدیرعامل و مسئول فنی. |
| فهرست مواد اولیه (BOM) | الزامی | الزامی | باید شامل تمام مواد و تأمینکنندگان باشد. |
| نمودار فرآیند عملیات (OPC) | الزامی | الزامی | نقشه بصری جریان تولید. |
| برچسبگذاری و دستورالعمل استفاده | الزامی | الزامی | باید با ضوابط ملی برچسبگذاری مطابقت داشته باشد. |
| پرونده فنی کامل | تعهد به تهیه و نگهداری | الزامی برای ارسال و بررسی | شامل فایلهای طراحی، تحلیل ریسک، صحهگذاری و…. این یک تمایز عمده بین کلاسها است. |
| اعتبارسنجی و صحهگذاری | |||
| گزارشهای آزمون آزمایشگاهی | بر اساس نیاز اداره کل | الزامی | از آزمایشگاههای معتبر IMED برای اثبات ایمنی و عملکرد. |
| گزارش ارزیابی بالینی (CER) | به ندرت مورد نیاز است | الزامی (به ویژه برای C و D) | شواهد حیاتی برای اثبات سودمندی و ایمنی بالینی. |
| تأسیسات و کدها | |||
| انطباق با GMP (عکس/فیلم/ممیزی) | الزامی | الزامی | اثبات وجود محیط تولید مناسب. |
| کد GTIN | الزامی | الزامی | پیشنیاز دریافت IRC و قابلیت ردیابی. |
بخش ۵: استانداردهای تأسیسات و عملیات: فراتر از کاغذبازی
اخذ پروانه ساخت تنها به ارائه مستندات کامل محدود نمیشود؛ تولیدکننده باید ثابت کند که دارای یک محیط تولیدی مناسب و فرآیندهای عملیاتی کنترلشده است.
اصول تولید خوب (GMP) در چارچوب ایران
رعایت اصول GMP برای تضمین کیفیت بالای تجهیزات پزشکی ضروری است. این اصول شامل الزامات دقیقی برای طراحی و نگهداری ساختمان و تأسیسات، کالیبراسیون و نگهداری تجهیزات تولید و آزمایش، بهداشت فردی کارکنان، و صحهگذاری فرآیندهای تولید است. انطباق با این اصول معمولاً در طی یک بازرسی حضوری از محل تولید توسط کارشناسان اداره کل یا دانشگاههای علوم پزشکی مربوطه ارزیابی میشود.
آمادگی برای ممیزی محل تولید
بازرسی از خط تولید یک مرحله حیاتی در فرآیند صدور پروانه است. برای آمادگی، شرکتها باید اطمینان حاصل کنند که:
- تمام مستندات (به ویژه پرونده فنی و سوابق QMS) به راحتی در دسترس هستند.
- خط تولید عملیاتی و تمیز است و فرآیندها دقیقاً مطابق با نمودار OPC ارائه شده انجام میشود.
- کارکنان کلیدی، به ویژه مسئول فنی و مدیر تولید، برای پاسخگویی به سؤالات فنی و رویهای ممیزان آماده هستند.
- برای کلاسهای خطر پایینتر، ارائه فیلم و عکس باکیفیت از خط تولید که تمام جزئیات فرآیند را نشان دهد، ممکن است به عنوان جایگزین یا مکمل بازرسی حضوری پذیرفته شود.

تعهدات نظارت پس از فروش (PMS) و قابلیت ردیابی
دریافت پروانه ساخت، پایان راه نیست، بلکه آغاز یک تعهد بلندمدت است. تولیدکننده از نظر قانونی موظف است عملکرد محصول خود را پس از ورود به بازار به طور مداوم رصد کند. این فرآیند که نظارت پس از فروش (PMS) نامیده میشود، شامل جمعآوری و تحلیل بازخوردها، گزارش حوادث ناگوار (Adverse Events) و رسیدگی به شکایات است. علاوه بر این، شرکت باید یک سیستم ردیابی مؤثر با استفاده از کدهای IRC و GTIN و شماره سریال یا لات ساخت (Lot Number) داشته باشد تا در صورت نیاز به فراخوان (Recall) محصول از بازار، بتواند به سرعت و به طور کامل اقدام کند.
بخش ۶: ارزش استراتژیک پروانه ساخت
پروانه ساخت، بیش از یک الزام قانونی، یک دارایی استراتژیک قدرتمند است که مزایای تجاری قابل توجهی را برای تولیدکنندگان به ارمغان میآورد.
- گشایش دسترسی به بازار و امکانپذیری تجاری: این پروانه، کلید ورود به بازار چند میلیارد دلاری تجهیزات پزشکی ایران است. بدون آن، فروش محصول به هر شکلی غیرقانونی است. با داشتن پروانه، تولیدکنندگان میتوانند محصولات خود را به طیف وسیعی از مشتریان، از جمله بیمارستانهای دولتی، کلینیکهای خصوصی، و داروخانهها عرضه کنند.
- ایجاد اعتبار و اعتماد: پروانه ساخت IMED به منزله یک مهر تأیید رسمی است که به پزشکان، مدیران بیمارستانها و بیماران اطمینان میدهد که محصول مورد نظر، استانداردهای دقیق ایمنی و کیفیت را گذرانده است. این اعتبار، یک ابزار بازاریابی قدرتمند است که به ایجاد تمایز رقابتی و جلب اعتماد مشتریان کمک میکند.
- پیشنیاز شرکت در مناقصات دولتی و فرصتهای گستردهتر: بیمارستانها و مراکز درمانی دولتی، که بخش بزرگی از بازار را تشکیل میدهند، تنها مجاز به خرید تجهیزات پزشکی دارای پروانه ساخت و کد IRC هستند. این مجوز همچنین یک عامل کلیدی در جذب سرمایهگذار و دریافت تسهیلات بانکی است، زیرا نشاندهنده پایداری و قانونی بودن کسبوکار است.
- تسهیل صادرات و ورود به بازارهای بینالمللی: اگرچه پروانه ساخت یک مجوز ملی است، اما فرآیند دقیق اخذ آن، به ویژه الزام به انطباق با استاندارد جهانی $ISO 13485$، یک زیرساخت کیفیتی مستحکم برای شرکت ایجاد میکند. این زیرساخت، مسیر اخذ گواهینامههای بینالمللی مانند نشان CE را بسیار هموارتر کرده و به شرکت در ورود به بازارهای صادراتی کمک شایانی میکند.
- تقویت صنعت داخلی و کاهش وابستگی به واردات: دولت از طریق سیاستهای حمایتی، از تولیدکنندگان داخلی دارای پروانه ساخت پشتیبانی میکند. این سیاستها ممکن است شامل محدودیت واردات محصولات مشابه خارجی باشد که این امر یک مزیت رقابتی و یک بازار محافظتشده برای تولیدکنندگان بومی ایجاد میکند.

بخش ۷: پیمایش فرآیند: دیدگاههای تخصصی و اجتناب از اشتباهات رایج
فرآیند اخذ پروانه ساخت مملو از جزئیات فنی و اداری است که نادیده گرفتن آنها میتواند منجر به تأخیرهای طولانی و هزینههای اضافی شود. آگاهی از اشتباهات رایج، اولین گام برای اجتناب از آنهاست.
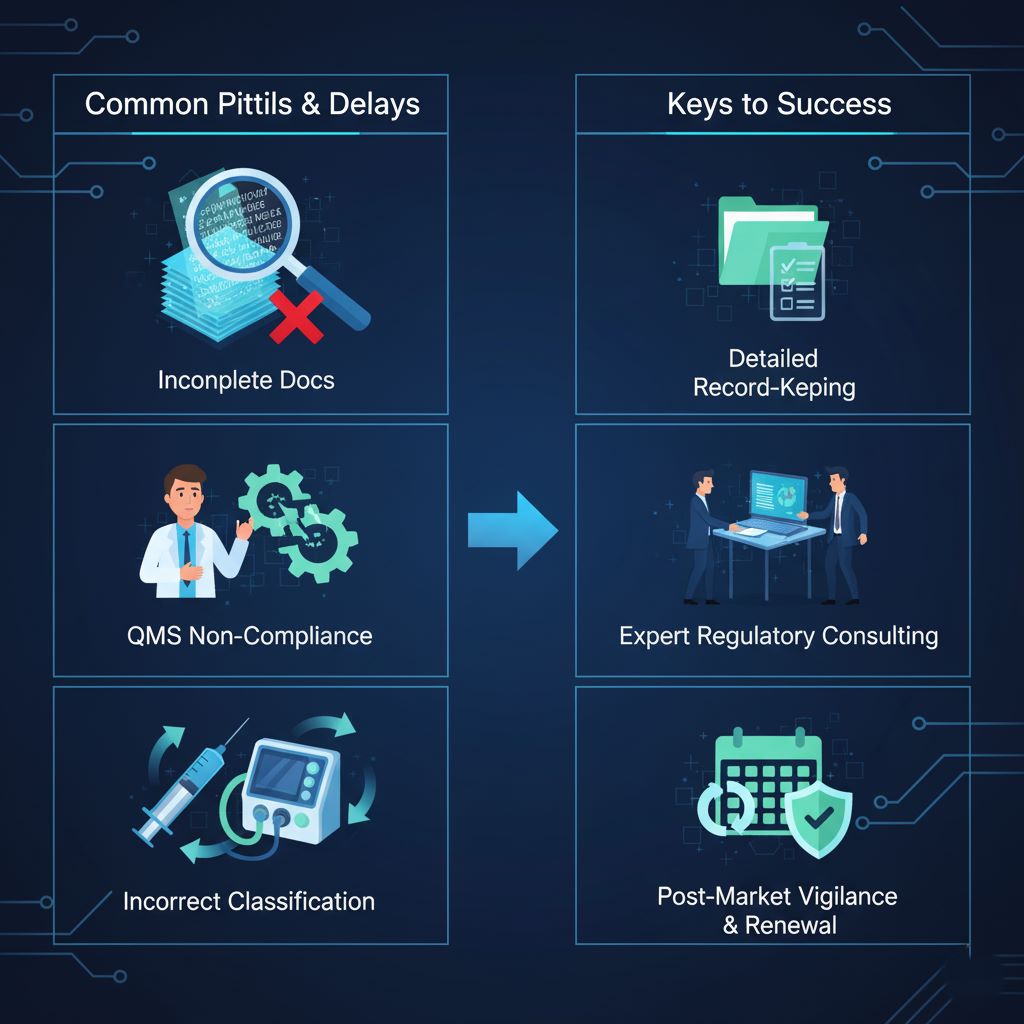
شایعترین دلایل رد یا تأخیر درخواست
- مستندات ناقص یا متناقض: این مورد، بزرگترین و شایعترین دلیل تأخیر در پروندهها است. ارائه پروندهای که در آن، اطلاعات اظهارنامه انطباق با نتایج گزارشهای آزمون همخوانی ندارد یا لیست مواد اولیه کامل نیست، به سرعت توسط کارشناسان شناسایی شده و منجر به درخواست رفع نقص میشود.
- دستکم گرفتن الزامات QMS/$ISO 13485$: بسیاری از شرکتها گواهینامه ایزو را صرفاً یک “کاغذ” میدانند که باید خریداری شود، در حالی که ممیزان به دنبال شواهد عینی از پیادهسازی واقعی یک سیستم مدیریت کیفیت کارآمد در تمام سطوح سازمان هستند. عدم وجود سوابق کافی و رویههای مستند، یک نقص عمده (Major Non-conformity) محسوب میشود.
- طبقهبندی نادرست محصول: اشتباه در تعیین کلاس خطر میتواند منجر به ارائه مجموعه اشتباهی از مستندات شود. به عنوان مثال، ثبت یک وسیله کلاس B با الزامات کلاس A، منجر به رد فوری درخواست و نیاز به شروع مجدد فرآیند خواهد شد.
- نادیده گرفتن تعهدات پس از فروش: عدم درک و برنامهریزی برای مسئولیتهای مداوم مانند PMS، گزارشدهی حوادث و آمادگی برای ممیزیهای مراقبتی، میتواند اعتبار پروانه را حتی پس از صدور به خطر اندازد.
اهمیت نگهداری سوابق دقیق
هر ادعایی که در پرونده فنی مطرح میشود، باید با شواهد مستند، قابل ردیابی و قابل تأیید پشتیبانی شود. اصل اساسی در ممیزی این است: “اگر مستند نشده باشد، انجام نشده است.” نگهداری سوابق دقیق از طراحی، تولید، کنترل کیفی و بازخورد مشتریان، ستون فقرات یک سیستم مدیریتی قوی است.
مشاوره با متخصصان نظارتی: چه زمانی و چرا یک سرمایهگذاری هوشمندانه است؟
با توجه به پیچیدگی قوانین و فرآیندها، استفاده از خدمات مشاوران متخصص در امور نظارتی میتواند یک سرمایهگذاری ارزشمند باشد. این متخصصان با تسلط بر آخرین دستورالعملها و رویههای داخلی اداره کل، میتوانند از بروز اشتباهات پرهزینه جلوگیری کرده، زمان رسیدن به نتیجه را به طور قابل توجهی کاهش دهند و به شرکت در تدوین یک استراتژی نظارتی مؤثر کمک کنند.
درک چشمانداز پس از تأیید: ممیزیها، تمدید و حفظ انطباق
پروانه ساخت معمولاً برای یک دوره ۵ ساله صادر میشود و اعتبار آن منوط به موفقیت در ممیزیهای مراقبتی دورهای است که حداقل سالی یک بار انجام میشود. شرکتها باید همواره سیستم مدیریت کیفیت خود را در حالت “آماده برای ممیزی” نگه دارند. فرآیند تمدید پروانه نیز نیازمند ارائه مستنداتی است که نشاندهنده حفظ انطباق و عملکرد مناسب محصول در طول دوره اعتبار پروانه است.
نتیجهگیری
مسیر دریافت پروانه ساخت تجهیزات پزشکی در ایران، یک فرآیند دقیق، ساختاریافته و چالشبرانگیز است که نیازمند تعهد کامل به کیفیت و انطباق با مقررات است. این سفر با ایجاد یک زیربنای قانونی و حقوقی مستحکم آغاز میشود، با پیادهسازی یک سیستم مدیریت کیفیت جامع بر اساس استاندارد $ISO 13485$ ادامه مییابد، و با تدوین یک پرونده فنی بینقص و اثبات ایمنی و عملکرد محصول به اوج خود میرسد. هر مرحله از این مسیر، از طبقهبندی صحیح محصول تا آمادگی برای ممیزیهای حضوری، نقشی حیاتی در موفقیت نهایی ایفا میکند.
در نهایت، شرکتهایی در این عرصه موفق خواهند بود که به این فرآیند نه به عنوان یک مانع بوروکراتیک، بلکه به عنوان یک مزیت رقابتی استراتژیک نگاه کنند. رعایت دقیق این مقررات، تنها هزینه ورود به بازار نیست؛ بلکه یک سرمایهگذاری بلندمدت در کیفیت، ایمنی و اعتبار است. این سرمایهگذاری، زیربنای یک کسبوکار پایدار، قابل اعتماد و در نهایت، سودآور را در صنعت پویای تجهیزات پزشکی ایران بنا مینهد.
لینکهای مهم اوکی صنعت